热线:021-66110810,66110819
手机:13564362870
热线:021-66110810,66110819
手机:13564362870
摘要:了解FDA和EMA参比制剂校正平均生物等效性(RSABE)方法的区别及具体应用,为行业人员提供方法学的指导。探讨高变异药物重复交叉设计的参比制剂校正平均生物等效性方法及选择原则,通过模拟研究和实例分析对FDA和EMA方法在不同情况下的结果表现进行比较。当个体内变异≤30%或样本量较大时,FDA和EMA方法生物等效通过率基本一致;在个体内变异40%-70%,且样本量较低时,FDA方法生物等效通过率要高于EMA。在样本量较大的重复交叉设计中,EMA和FDA提出的参比制剂校正平均生物等效性方法均有良好的表现,但EMA对样本量要求更高,FDA的几何均值比限制条件必不可少。
高变异药物(highly variable drug)是指生物等效(bioequivalence,BE)评价指标药动学参数(AUC0-1,AUC0-∞,及Cmax)的个体内变异系数(with-in-subject coefficient of variation,CV)≥30%的药物。对于这类药物,采用常规18~24例2 X 2交叉设计和等效界限(bioequivalence limits,BEL)进行生物等效性评价时,由于个体内变异增大,使检验把握度降低,极可能犯统计学上的Ⅱ类错误,造成的结果是将实际与参比制剂生物等效的受试制剂判断为不等效[1]。2006年,FDA向药学咨询委员会提出了适用于高变异药物的参比制剂校正平均生物等效性(reference-scaled average bioequivalence,RSABE)的方法。2010年,EMA发布了生物等效性研究指导原则[2],也提出了RSABE方法。FDA与EMA提出的RSABE方法,在试验设计、校正理论方面是一致的,但在生物等效性判定界限及程序实现上是有区别的[3]。
我国对于高变异药物生物等效研究通常是通过增大样本量来达到等效的目的。2007年国家食品药品监督管理总局药品审评中心(CDE)电子刊物发表文章"关于高变异药物生物等效性研究的考虑"提到了"比例标化平均生物等效性"的方法[4]。2015年国家药典委员会发布的《药物制剂人体生物利用度和生物等效性试验指导原则》中,关于高变异药物也可以采用重复交叉设计对药动学参数指标BEL进行放宽,Cmax的BEL最宽为69.84%~143.19%,这与EMA关于RSABE方法校正的最宽界限一致[5]。
国内可见介绍RSABE理论的文献报道[3,6],本研究主要通过模拟研究和实例分析来对比FDA和EMA提出的RSABE方法,旨在为相关从业人员提供方法学的指导。
1、试验设计
RSABE方法需要应用参比制剂的个体内变异,因此参比制剂需要至少使用2个周期,重复试验设计,包括以下2种类型。
1.1、半重复、三周期交叉实验设计
将试验受试者随机分为两组,两组受试者3个周期的用药顺序为TRR/RRT/RTR(R表示参比制剂,T表示试验制剂)。
1.2、全重复,四周期交叉试验设计
将试验受试者随机分为两组,两组受试者4个周期的用药顺序为TRTR/RTRT。
2、样本量确定
Laszlo等[7]在2011年发表的关于高变异药物生物等效试验设计样本量的文章中,分别对3周期或4周期交叉设计试验,按照不同的个体内变异、检验效能、几何均值比以及FDA和EMA不同的限制性要求,给出了样本量的模拟计算结果。Reddy等[8]采用公式近似法给出了不同参数设置下的样本量情况。刘甜甜等[9]应用确切样本量计算方法估算了高变异药物生物等效研究所需的样本量。以上研究结果均显示,对于高变异药物,同样的估计参数,4周期交叉设计样本量要比3周期交叉设计低30%,FDA要求的样本量要低于EMA。
3、RSABE方法介绍
生物等效试验中,两制剂药动学参数指标如果等效,常采用平均生物等效(average bioequivalence,ABE)方法,需满足如下条件:
(μT-μR)2≤θA2式(1)
μT和μR表示试验制剂和参比制剂药代参数指标对数转换均值;θA取值为In(1.25),式(1)可变换为:In(0.8)≤μT-μR≤In(1.25)或0.8≤eμT/eμR≤1.25。如果两制剂药动学参数指标几何均值比(geometric mean ratio,GMR)的90%置信区间(confidence interval,CI)在等效界限(0.8~1.25)内,则认为两制剂等效。
与ABE相比,若采用RSABE方法,需采用重复交叉试验设计,药动学参数指标如果等效,需满足:

进行生物等效性判定,即RSABE。FDA和EMA采用RSABE和ABE方法的选择原则见图1[10]。
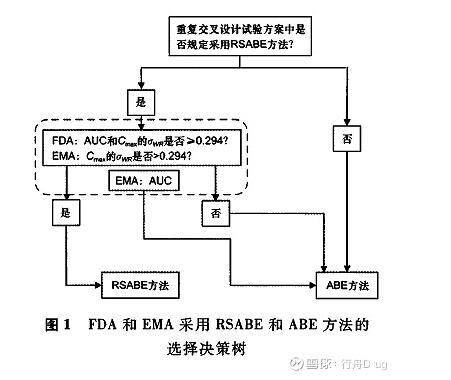
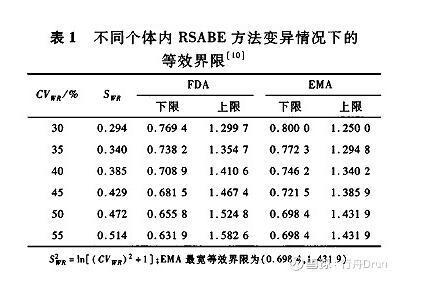
FDA将σW0常数设为0.25,EMA设为0.294。在实际应用时,FDA按照药动学参数个体内标准差>0.294(个体内变异30%),即采用RSABE方法,因此FDA的GMR 90%置信区间的等效界限是不连续的,而EMA是连续的[11]。RSABE方法不同个体内变异情况下的等效界限见表1。
4、RSABE应用步骤
步骤1:计算参比制剂个体内标准差(SWR)。
FDA:试验设计采用半重复和全重复交叉设计,SWR计算公式为:


5、模拟研究
为考察不同个体内变异、不同样本量及不同几何均值比,FDA和EMA方法的BE通过情况,本研究使用统计分析软件(SAS 9.4)模拟生成3周期交叉设计,对数转换Cmax数据。该数据服从多元正态分布,周期间相关系数假设为0.4。设受试制剂和参比制剂个体内变异相同,分别为20%,30%,40%,50%及70%,样本量分别为24,48,72,GMR设定从1.0到1.6,共生成模拟数据集105个,每个数据集模拟分析1,000次,分析方法采用FDA,EMA和FDAnc(nc:no GMR constraint,即无GMR限制条件),模拟分析结果见图2。
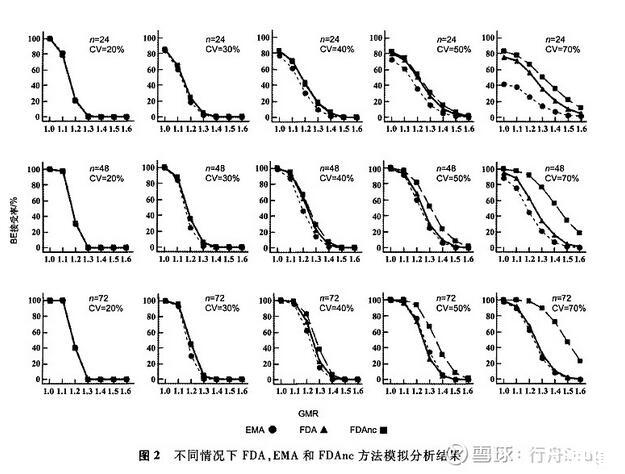
模拟结果显示:样本量越大,相同变异情况下2种方法的BE通过率越高。个体内变异为20%时,EMA和FDA方法BE通过率曲线完全重叠。个体内变异为30%,理论GMR设定在1.1~1.3时,FDA方法BE通过率略高于EMA,但差别不大。随着变异增大,FDA方法显示出比EMA更高的BE通过率,尤其在样本量较低时更为明显。此外,在变异较大时(50%~70%),FDAnc由于不对GMR添加限制,其BE通过率远大于FDA和EMA方法,此差异随着样本量增加而越发突出。在样本量较大(72例),变异相同情况下,FDA和EMA方法BE通过率基本一致。
6、实例分析
某国产药物为片剂,参比为进口原研制剂,需进行空腹及餐后BE研究。试验设计时,查阅国外同品种药物研究及相关文献均表明该药物为高变异药物。因此,设计时采用半重复3 X 3交叉设计,样本量按照个体内变异>30%,GMR在0.9-1.1,并考虑脱落情况,空腹及餐后各计划筛选入组57例受试者,实际空腹完成3周期给药52例,餐后49例。试验结束后,使用WinNonlin®软件计算药动学参数,并整理数据为表2。
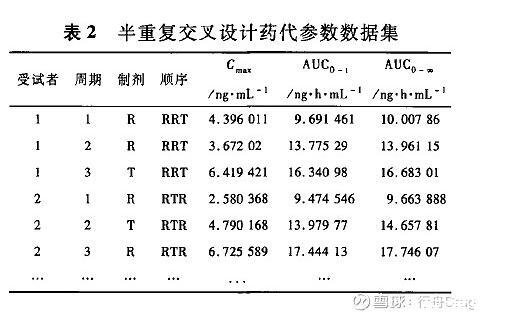
按照试验方案规定,本研究满足条件时可采用RSABE方法进行生物等效判定。依据FDA和EMA原则,本研究使用SAS 9.4分析软件编程进行生物等效分析,分析结果见表3和表4。
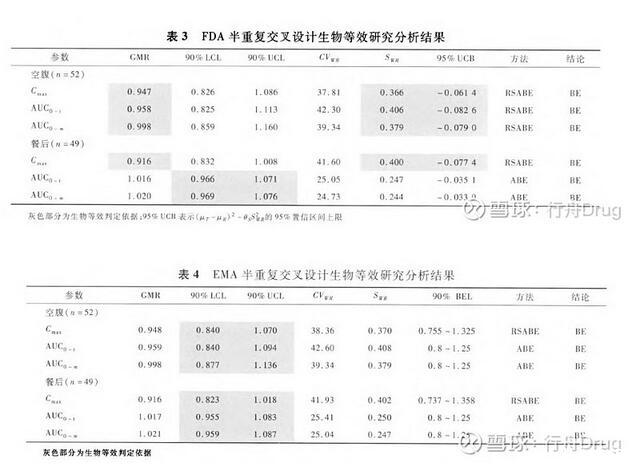
分析结果显示:FDA原则下,空腹试验的药动学参数指标及餐后试验的Cmax个体内标准差均>0.294,采用RSABE方法,这些药动学参数指标95%UCB均小于0,且GMR点估计值在0.8~1.25界限内。餐后试验AUC指标采用ABE估计90%置信区间,均在0.8~1.25等效界限内。综上可认为两制剂生物等效。依据EMA原则,最终分析结论一致。但EMA只在Cmax个体内标准差>0.294时,采用RSABE方法,AUC指标仍使用ABE方法。此外,从置信区间来看,EMA和FDA的差别不大。
7、结语
重复交叉设计常用于高变异药物生物等效性研究,相比双交叉设计可以节省一定的样本量,但由于给药周期多,如果再有较长的洗脱期,受试者的脱落率可能会成倍增加。因此在决定是否采用重复交叉设计时,首先应考虑药物自身特点,是否高变异、半衰期长短、是否有后遗效应等;其次在方案设计时需要与药监部门进行充分的咨询与沟通,以保证试验设计的科学性和可行性。
在重复交叉设计生物等效研究中,当个体内变异小于30%时,FDA和EMA的等效界限均为0.8~1.25。当高于30%时,FDA会随着个体内变异增大,等效界限逐渐放宽。而EMA对等效界限的放宽仅限于变异≤50%。在相同的个体内变异情况下,FDA校正的等效界限要宽于EMA,这也决定了在样本量估计时,EMA的要求更高。模拟研究也证实,变异>30%时,EMA的BE通过率要低于FDA,因此要达到同样的效能,EMA需要更多的样本量。
随着样本量的增加,2种方法的BE通过率都有提升,但是对EMA方法的提升更加突出。FDA的RSABE等效判定时,GMR点估计值在0.8~1.25是重要的限制条件,模拟研究表明,尤其当变>50%,样本量较大时,FDAnc方法的BE通过率要远大于EMA和FDA方法,这就可能出现两制剂均值差异达50%,但结论却等效的情况。因此,GMR限制条件在FDA原则下是必不可少的[11,16]。从模拟研究结果可以看到,当个体内变异≤30%或样本量较大时,FDA和EMA方法基本一致,而当个体内变异在40%~70%,且样本量较低时,FDA方法BE通过率要高于EMA。因此,在评价高变异药物生物等效时,无论采用FDA还是EMA方法,具备一定的样本量是生物等效研究成功的前提[17]。
参考文献作者:

 相关新闻
相关新闻