热线:021-66110810,66110819
手机:13564362870
热线:021-66110810,66110819
手机:13564362870
当前证据表明,硫化氢(H₂S)在脑功能中扮演重要角色,可能作为神经调质和细胞内信使发挥作用。在哺乳动物中枢神经系统(CNS)中,H₂S由氨基酸半胱氨酸通过胱硫醚β-合成酶(CBS)催化生成,同时产生副产物丝氨酸(Ser)。由于CBS是一种钙离子和钙调蛋白依赖性酶,H₂S的生物合成应受细胞内钙离子浓度的急性调控。此外,S-腺苷甲硫氨酸(SAM)作为CBS的变构激活剂也参与调控。
过去十年在阐明H₂S在生理和病理条件下的细胞水平作用方面取得重大进展。生理水平的H₂S首次被证明选择性刺激NMDA受体介导的电流。这种刺激促进海马LTP诱导,但仅在弱强直刺激存在下。H₂S单独不诱导LTP,表明H₂S主要在活跃突触促进LTP(Abe and Kimura,1996;Kimura,2000)。H₂S增强NMDA受体功能的潜在机制尚不清楚,尽管一种可能的途径是通过氧化还原调节散布于神经元NMDA受体胞外域的巯基,这些巯基对氧化/还原剂敏感。许多内源性(如吡咯喹啉醌、硫辛酸、活性氧自由基、谷胱甘肽、二氢硫辛酸)和外源性分子(氰化物、氟吡汀)能够氧化和还原NMDA受体(Dingledine et al.,1999),分别导致受体反应减弱和增强。因此,H₂S可能凭借其还原特性激活NMDA受体。一个可能的氧化还原调节位点是位于NR1亚基胞外域的Cys对(Cys744和Cys798)(Sullivan et al.,1994)。
在细胞内,H₂S通过cAMP生成增强NMDA受体介导的反应。外源性H₂S增加大鼠原代大脑和小脑神经元培养物或某些神经元和胶质细胞系中cAMP生成(Kimura,2000)。cAMP在LTP启动和晚期激活cAMP依赖性蛋白激酶(PKA)(Roberson and Sweatt,1996;Abel et al.,1997)。激活的PKA可能进而磷酸化NMDA受体亚基NR1、NR2A和NR2B的特定位点,以增强NMDA电流(这对LTP诱导至关重要)(Leonard and Hell,1997;Tingley et al.,1997)。因此,cAMP可能通过磷酸化NMDA受体调节LTP。除此之外,H₂S还通过cAMP途径以剂量依赖性方式减少对NMDA作出反应所需时间,即增加NMDA受体对其配体的敏感性。
最近还显示,H₂S上调γ-氨基丁酸(GABA)B受体(GABABR),这是一种位于突触前和突触后位点的G蛋白偶联受体(Han et al.,2005)。刺激突触后受体产生长时程抑制性突触后电位,导致K⁺电导增加,对抑制性神经传递的微调很重要。H₂S已显示通过增加K⁺外流(可能通过ATP依赖性K⁺(KATP)通道)使CA1区和中缝背核神经元超极化(Reiffenstein et al.,1992)。在突触前位点,GABABR通过抑制电压敏感性Ca²⁺通道调节神经递质(如GABA和Glu)释放。H₂S上调GABABR表达暗示H₂S可能在维持脑内兴奋/抑制平衡中起作用。
除其神经调节作用外,H₂S已被证明在细胞外和细胞内微环境中保护神经元免受氧化应激。公认还原型谷胱甘肽(GSH)是脑中重要的抗氧化防御。它通过清除自由基和其他活性物质、去除过氧化氢和脂质过氧化物、防止生物分子氧化来保护大脑(Wu et al.,2004综述)。H₂S与GSH具有相似的神经保护特性,在体外具有相当的效力。这已通过H₂S的能力得到证明:(i)抑制次氯酸介导的氧化损伤(Whiteman et al.,2005);(ii)抑制过氧亚硝酸盐介导的蛋白质硝化和细胞毒性(Whiteman et al.,2004)。此外,H₂S在体外轻易清除过氧化氢(H₂O₂)(Geng et al.,2004),后者是大多数细胞氧化应激的重要来源。尽管神经元(和胶质细胞)内谷胱甘肽水平在毫摩尔浓度范围,但脑内其细胞外水平几乎为零(Halliwell,2001;Bayir et al.,2002;Whiteman et al.,2005)。因此,细胞外环境高度依赖其他稳态调节的非谷胱甘肽抗氧化剂(如抗坏血酸)清除自由基(Rice,2000)。因此,由于其高内源性生成、易扩散特性和与GSH相当的强抗氧化效力,H₂S可能成为脑细胞外微环境中另一种重要的内源性抗氧化剂候选物。
H₂S增加神经元中还原型谷胱甘肽(GSH)的生成(Kimura and Kimura,2004)。NaHS处理本身能够通过增强γ-谷氨酰半胱氨酸合成酶(γ-GCS)活性和上调Cys(GSH合成的限速底物)转运来增加谷胱甘肽量。这种谷胱甘肽含量的增加被证明保护神经元免受氧毒性(一种由氧化应激引发的程序性细胞死亡),后者由高浓度Glu触发。H₂S还通过激活ATP依赖性K⁺(KATP)和Cl⁻通道以及增加谷胱甘肽水平,保护永生化小鼠海马细胞系细胞免受氧化性Glu毒性。通过这两种机制,H₂S能够在不同类型的神经细胞中提供对Glu诱导细胞死亡的完全保护,将细胞活力提高至未用Glu处理的神经元相似水平(Kimura and Kimura,2004;Kimura et al.,2006)。
胶质细胞
H₂S在胶质细胞中扮演重要的神经调节作用。星形胶质细胞作为胶质细胞的主要类型,在维持神经元兴奋性、调节脑pH稳态以及摄取突触周围的多种神经递质(包括谷氨酸)中发挥关键作用(Koehler et al.,2006)。更重要的是,星形胶质细胞内钙离子浓度([Ca²⁺]i)的充分增加能够诱导并传播至邻近星形胶质细胞的扩散性钙离子升高波,称为“钙波”(Dani et al.,1992)。与神经元通过产生动作电位传递信号不同,星形胶质细胞和其他胶质细胞通过钙信号相互通讯(Braet et al.,2004综述)。这为星形胶质细胞作为合胞体调节神经元和血管功能提供了基础,暗示胶质细胞在突触传递中的整体调节作用(Braet et al.,2004;Koehler et al.,2006)。外源性H₂S可在原代星形胶质细胞培养物和海马切片中引发钙波(Nagai et al.,2004)。这种由H₂S触发的钙波之前会伴随[Ca²⁺]i的升高,该升高通过质膜上的钙通道介导钙内流,并在较小程度上通过细胞内钙库的钙释放实现。在脑切片中,H₂S引起的[Ca²⁺]i增加会扩散至邻近星形胶质细胞群并触发钙波。
与小胶质细胞不同,小胶质细胞作为常驻中枢神经系统的巨噬细胞群体,在外来挑战时可被激活,类似于外周巨噬细胞(Farber and Kettenmann,2005综述)。小胶质细胞在阿尔茨海默病(AD)(Wojtera et al.,2005)和帕金森病(Kim and Joh,2006)等神经元疾病的进展中被认为发挥作用。我们近期发现,外源性H₂S应用可显著提高小胶质细胞的[Ca²⁺]i,且呈剂量依赖性(Lee et al.,2006)。外源性H₂S通过质膜介导钙内流以及细胞内钙库释放钙离子触发钙内流。这种内流部分依赖于腺苷酸环化酶的激活,且独立于磷脂酶C-蛋白激酶C-三磷酸肌醇通路。此外,抑制内源性H₂S合成显著降低[Ca²⁺]i,表明内源性H₂S可能对[Ca²⁺]i稳态具有正向调节作用。除作为第二信使外,钙离子作为整合因子控制小胶质细胞在静息和激活状态下的行为,其中基础钙浓度升高是脂多糖(LPS)刺激后激活小胶质细胞的特征(Hoffmann et al.,2003)。凭借H₂S的易扩散特性,可以合理推测H₂S可能通过升高邻近小胶质细胞的基础钙水平在激活它们中发挥作用。
神经炎症是一种由促炎细胞因子介导的过程,可由全身组织损伤引发,但最常见的是与神经系统直接损伤相关。神经炎症涉及激活免疫细胞、胶质细胞和神经元的神经-免疫相互作用(Myers et al.,2006)。目前认识到所有主要的神经病理状态均以显著的炎症反应为特征,这些反应主要由胶质细胞介导。激活的胶质细胞产生各种促炎或抗炎趋化因子,被认为对启动和引导免疫细胞浸润至脑组织并协调其活动至关重要(Morale et al.,2006)。小胶质细胞是驻留于中枢神经系统的特化巨噬细胞,其在神经炎症中的激活作用至关重要(Moore and O'Banion,2002;Craft et al.,2005)。激活的小胶质细胞产生并释放某些促炎因子(如一氧化氮、肿瘤坏死因子-α(TNF-α)和白介素-1β(IL-1β)),这些因子进一步加剧组织损伤并导致细胞死亡(Wojtera et al.,2005)。Hu等(2007)近期报道,H₂S通过显著减弱LPS刺激的胶质细胞中一氧化氮和TNF-α分泌,以浓度依赖性方式保护小胶质细胞和星形胶质细胞免受LPS诱导的炎症反应。其机制似乎涉及抑制LPS诱导的诱导型一氧化氮合酶(iNOS)表达和p38丝裂原活化蛋白激酶(MAPK)磷酸化。
尽管有证据表明H₂S对神经元具有神经保护作用,但迄今为止尚未开展关于H₂S对胶质细胞群体保护作用的研究。多项研究证明谷胱甘肽(GSH)优先定位于胶质细胞(Slivka et al.,1987;Raps et al.,1989),胶质细胞中平均细胞内水平为4 mM,而神经元中为2.5 mM(Rice and Russo-Menna,1998)。鉴于H₂S在神经元中增加GSH含量的作用,H₂S可能对胶质细胞发挥相似甚至更强的效应以增强抗氧化性GSH。
生理功能
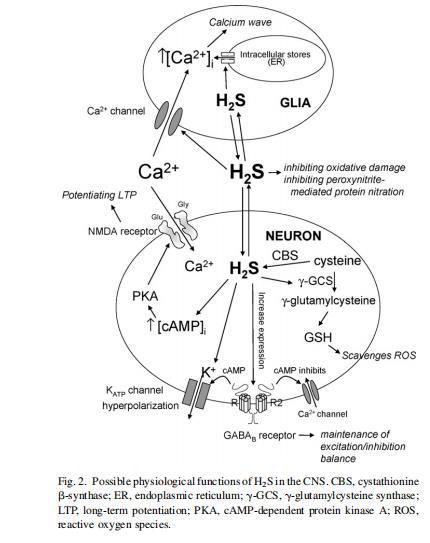
基于上述H₂S的已知作用,其在脑中的潜在生理功能可能包括钙稳态调节、LTP增强、氧化应激抑制以及神经传递调控(图2)。
