热线:021-66110819,13564362870
Email:info@vizai.cn
热线:021-66110819,13564362870
Email:info@vizai.cn
光生物制氢效果很好 其重要性,因为它有望产生清洁的可再生能源。 在自然界中,绿藻不能产生氢气,因为 氢化酶对氧气极度敏感的结果。 然而,我们发现硅化诱导的绿藻 聚集体可以实现可持续的光生物氢 即使在自然有氧条件下也能生产。 核心- 绿藻聚集体的壳结构创造了一种平衡 光合电子产生和氢化酶之间 活性,从而允许产生氢气。 这一发现 为太阳能驱动的水分解提供了可行的途径 转化为氢气和氧气以开发绿色能源替代品 通过使用合理设计的细胞-材料复合物。
氢气(H2 ) 被认为是一个有前途的替代品 对于化石燃料,由于其卓越的转换效率, 环保,高能量容量。 [1] H2 今天的生产主要依赖蒸汽 碳氢化合物重整、煤气化和 核动力水电解,这是能源密集型和不可持续的。 [2] 太阳能生物制氢 电源提供了产生 H2 的可能性 那是可再生的 和碳中和,因为它直接使用取之不尽的资源:太阳能和来自 H2O 的电子。 [3] 在自然界, 光合微生物,特别是绿藻,可以 使用氢化酶(一种酶 催化分子氢的可逆氧化) 耦合到光合作用机器。 [4] 然而,这是 在几分钟内发生的短暂过程 暗光过渡。 [5] 这是因为氢化酶失去了它的 在有氧的情况下发挥作用。 [6] 在黑暗中,细胞呼吸产生激活氢化酶的厌氧条件。 [7] 在从黑暗走向光明的瞬间, 来自光系统 II (PSII) 反应中心的水光氧化反应 (H2O!2H+ + 1/2O2 + 2 e¢ ) 的光合电子可以传递给氢化酶以产生 H2 (2H+ + 2 e¢ !H2)。 然而 光合作用反应迅速产生氧气 使氢化酶失活。 [8] 为了提高光生物 H2 产量,科学家筛选了 [9a,b] 和 构建了具有改善的氧耐受性的突变体和 通过降低内在氢化酶氧敏感性 各种策略。 [9c] 不幸的是,进展有限 制作。 目前,硫剥夺是最常见的 用于产生绿色厌氧环境的方法 藻类。 [7a] 然而,这种代谢处理可以同时抑制 PSII 活性并逐渐终止随后的 H2 产生。 [7a, 10] 由于不利结果,绿藻在 H2 中的大规模应用 生产仍然不现实。
在自然界中,许多生物体,如硅藻, 球石和趋磁细菌已经从生物矿化过程中发展出特定的矿物结构 提供广泛的保护和独特的功能。 [11] 近期 研究成果表明,仿生矿化可以 作为一种有用的工具来改造细胞和病毒,以及 由此产生的细胞-材料复合物总是有不同的 来自原生生物的生物学特性。 [12] 这里我们 报告一种可以赋予绿藻的仿生硅化 具有可持续光生物 H2 的新能力 在自然大气条件下生产。
蛋白核小球藻 (C. pyrenoidosa) 是一种单细胞 绿藻在其叶绿体中具有突出的核糖体, 也是少数商业化的微藻物种之一 已被大规模培养。 通常,细胞是 在单细胞状态下,直径 3-4 毫米,没有倾向 自聚合(图 1a)。 聚(二烯丙基二甲基氯化铵)(PDADMAC) 分子可以模拟硅化蛋白诱导 原位二氧化硅沉积在细胞表面, [12b, e] 这可以 也适用于绿藻。 仿生后 修改后,硅化的 C. pyrenoidosa 细胞可以自聚集(图 1b)。 因此,硅化细胞具有 降低的 zeta 电位(约 ¢ 3 mV)与 原生细胞(约 ¢ 15 mV),这有利于 粒子团聚(支持信息,图 S1)。 我们还确认细胞通过 无定形SiO2 (支持信息,图S2)形成 细胞材料在硅化过程中聚集。 这 通过调节细胞硅化中的 C. pyrenoidosa 密度可以将聚集体尺寸控制在 10-500 mm 中(支持信息,图 S3 和 S4)。

图 1. 蛋白核小球藻细胞及其聚集体。 a) 本地人 使用光学显微镜和扫描电子观察细胞 显微镜(SEM)。 比例尺:20 毫米(插图:2 毫米)。 b) 聚合 使用光学显微镜和 SEM 观察细胞。 比例尺: 50 毫米(插图:2 毫米)。 c) 含有 WO3 的天然小球藻培养基 粉末(管的底部)。 d) 本地和聚合的小球藻培养物 管中装有 WO3 粉末的介质。 e) 聚合小球藻 含有 WO3 粉末的培养基(管底)。 酒吧 表示 WO3 的标准颜色变化 在 H2 存在下 在 解决方案。
一些金属氧化物的变色性质 它们的氧化态已被用于方便的光学 H2 检测。 [13] 氧化钨 (WO3) 是常用的,因为它 与 H2 反应 生产钨青铜,其颜色由浅黄色变为蓝灰色。 [13] 这里将 WO3 粉末添加到细胞培养物中作为 环境大气下 H2 产量的指标。 这 天然和聚集的 C. pyrenoidosa(约 100 毫米)的 H2 生产能力如图 1 c-e 所示。 在这 实验中,C. pyrenoidosa(总细胞密度 培养基为 3.0 × 107 细胞mL¢ 1 ) 被照亮 在 100 mEm¢ 2 s¢ 1 的光强度下 12 小时。 在本地 小球藻,WO3 粉末颜色不变表示无 可检测 H2 在系统中(图 1c),与 绿藻不能产生 H2 的传统观点 在自然有氧条件下。 然而,在综合 小球藻,WO3 粉末的颜色变成蓝灰色 (图 1 e),在溶液中产生 H2 之后 浓度约 10 mmolL¢ 1 (支持信息, 图 S5)。 需要注意的是,培养基是 暴露在空气中,因此产生的 H2 聚集的细胞发生在自然大气条件下。
为了定量研究聚集的小球藻细胞中 H2 产生的特性,我们监测了 H2 和 O2 在使用气体的密闭玻璃管的顶部空间中 色谱法(图 2a)。 含有小球藻 (1.2 × 108 细胞mL¢ 1 ) 加入 60 mL 光照下的密封玻璃管(100 mEm¢ 2 s¢ 1 )。 为了 原生小球藻,H2 无法检测到顶空 在任何测量点,顶部空间中的 O2 含量从最初的 21% 增加到 23%。 结果表明,天然小球藻细胞进行光合 O2 进化,但不产生 H2,这是由于 在有氧条件下使氢化酶失活。 有趣的是,观察到 100 毫米的聚集体以约 0.35 mmolH2h¢ 1 (mg叶绿素)¢ 1 (图 2a;支撑 信息,图 S6)。 这个比率是自然界瞬时生物质燃料产量的 1.75 倍 (0.20 mmolH2h¢ 1 (mg 叶绿素)¢ 1 ).[14] 更重要的是, 这种 H2 的产生不仅限于 几分钟; 相反,这种生产被检测到 至少 48 小时。 随后,H2 的速率 生产开始减少(支持信息, 图 S6),这可能是由于聚合受损 聚集体中细胞增殖产生的结构(支持信息,图 S7)。 的生产 在聚合之前,H2 可以持续至少 60 小时 结构分解(支持信息,图 S6 和 S7)。 与原生小球藻相比,我们注意到 O2 聚集体顶部空间的含量从 实验期间为 21% 至 19%。 这种减少很可能 归因于消耗 O2 分子的细胞呼吸 存在于封闭系统中。 在聚集的细胞中,O2 进化减少,因此 H2 产生 增加。 此外,在交替的时期 明暗(图 2 b),我们发现聚集体仅 在光照期间产生 H2; 下未检测到 H2 黑暗的条件。 这种明暗切换效应暗示着 H2 的产生与光合作用直接相关。
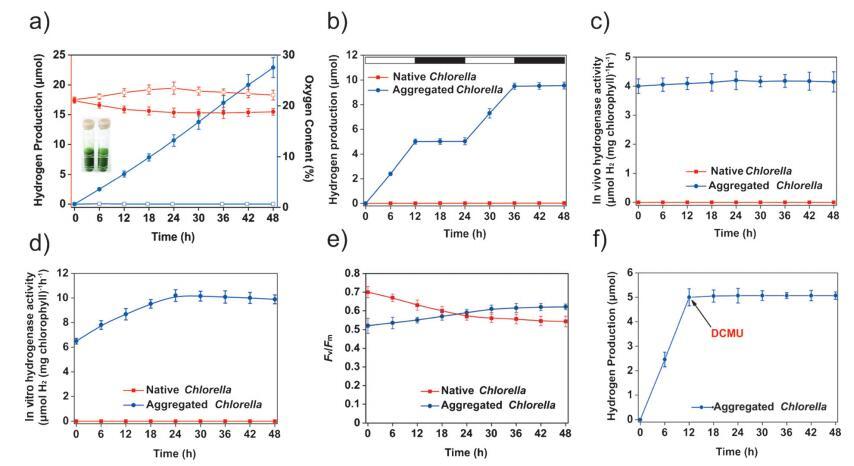
图 2. 在有氧条件下聚集的小球藻光生物制氢。 a) H2的量和O2的含量 在 100 mEm¢ 2 s¢ 1 光强下密封管的顶部空间 在不同的时间段(n=6)。 红线:O2%。 蓝线:H2amount。 空心方块:原生小球藻。 实心圆圈:聚集的小球藻。 b) 带有聚集小球藻的密封管中的累积 H2 积累 在明暗过渡期间(n = 5)。 c) 天然和聚集的小球藻 (n=5) 中的体内氢化酶活性。 d) 体外氢化酶活性 天然和聚集的小球藻 (n=5)。 e) 光强度下天然和聚集的小球藻 PSII (Fv/Fm) 的最大量子产率 100 mEm¢ 2 s¢ 1 在不同的时间段(n=3)。 f) DCMU 对聚集的小球藻 (n=3) 产生的 H2 的影响。
在绿藻中,光合水分解在功能上与活化的氢化酶产生的 H2 相关。 在 天然小球藻,既不是体内也不是体外氢化酶 可以检测到活性(图 2c 和 d),因为氢化酶在环境存在的情况下失活 氧气。 [6] 在小球藻聚集体中,体内氢化酶 活动在前 24 小时内略有增加,然后保持不变 在 4 mmolH2h¢ 1 的相对稳定水平 (mg叶绿素)¢ 1 (图 2 c),这也高于 H2 的速率 从大气中生产氩气(支持信息)。 该结果表明,氢化酶在 聚集体在大气氧水平下仍然活跃。 体外检查证实存在氢化酶 聚合中的活动,并且值甚至从 6.5 至 10.3 mmolH2h¢ 1 (mg 叶绿素)¢ 1 在前 24 小时内 (图2d)。 这些结果表明聚合 处理可能有利于氢化酶的表达 提高 H2 生产潜力。
除氢化酶外,影响持续光生物 H2 产生的另一个重要因素是其催化作用。 底物,即光合电子,起源于 来自 PSII 反应中的水氧化反应 中心。[15] PSII 是一种大的多亚基膜蛋白 可以捕获光子能量、分离电荷和 驱动类囊体膜中的电子转移。 [16] 该 PSII 的最大量子产率 (Fv/Fm) 已被广泛研究 用于评估 PSII 的活性。 Fv/Fm 的值在 实验开始时原生小球藻为 0.7, 并最终降至约 0.5(图 2e)。 这个 减少很可能是密封环境引起的应力的结果。 聚合中的 Fv/Fm 参数 小球藻在开始时减少到大约 0.5 实验(图 2 e),可能是由聚合处理引起的。 然而,最大量子产率 聚集的小球藻中的 PSII 从最初的 值 0.52 到最终值 0.62(图 2e)。 结果 表明适当的聚集处理几乎 不影响封闭细胞中 PSII 的活性,不像 硫剥夺治疗的影响。 [17] 化合物 3-(3,4-二氯苯基)-1,1-二甲基脲 (DCMU) 是 一种特定的 PSII 抑制剂,可破坏 从 QA 到 QB 的电子, [17] 及其添加到聚合 即使在光照条件下也完全终止制氢 (图2f)。 这一现象证实了 氢化酶与光合作用机器之间的耦合以产生 H2 在聚集的细胞中。
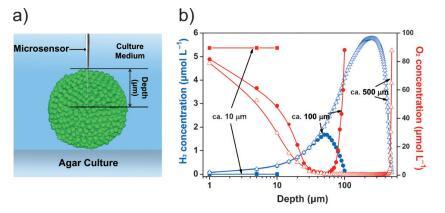
图 3. 聚集小球藻的空间功能分化。 a) 微传感器测量图。 b) 氢气 (蓝色)和 O2 (红色的) 不同大小的聚集小球藻的微观结构 (n=5)。
微传感器是研究生物的可靠工具 聚集体中的微环境(图 3a;支持信息,图 S8)。 在这里,我们使用 H2 和 O2 微型传感器来检查我们的聚集体中的内部环境。 在 100 毫米的骨料中,H2 的产量不 在骨料表面检测到。 H2浓度 随着探头深度的增加而增加,并达到 最大值(约 1.7 mmolL¢ 1 ) 在聚合核心 (图 3 b)。 此配置文件暗示 H2 是由核心产生的 细胞而不是表面细胞。 相比之下,O2 浓度随着探针深度的增加而降低。 这 核心微环境缺氧,O2 核心中心的浓度几乎为零,其中 氢化酶被激活,从而产生 H2。 聚合的空间 H2 和 O2 分布 小球藻细胞暗示存在空间功能 细胞之间的分化(SFD)。
聚合可以被认为是自生的 具有分化细胞功能的结构化核壳复合体。 表面细胞暴露在开放环境中,其功能与原生细胞相似 藻类。 然而,外面的细胞也起到了外壳的作用 防止环境 O2 渗透 进入聚合 核。 内细胞的呼吸反应消耗了任何 扩散的 O2 或光合作用产生的 O2 创造 缺氧域。 这种厌氧条件是由于 O2 扩散、PSII 产生的 O2 和隔离系统中的细胞呼吸之间的动态平衡由下式定义 封闭的外壳空间。 氢化酶和 PSII 活性 可以在这个域中保持,并且它们的耦合 保证可持续的光生物 H2 生产。 这 从个体绿藻到功能转变 聚集的绿藻可以通过 SFD 来理解 核壳结构:核心细胞产生氢气 由于隔离的厌氧微环境而产生的能力; 壳细胞保留了原生藻类的功能和 将核心细胞与开放环境隔离 (方案1)。
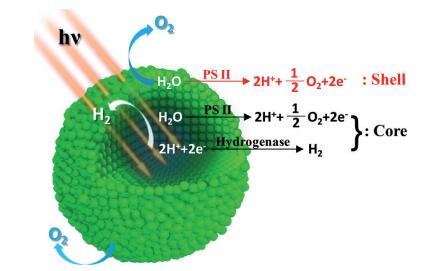
方案 1. 聚集小球藻的空间功能分化 细胞。
基于核壳的 SFD 对容量的影响 可以通过控制聚集体的大小来调整光生物 H2 的产生。 对于具有 直径约 10 毫米,H2 浓度在两个 可以忽略内部和外部单元格(图 3 b)。 O2 浓度保持在同一水平(约 90 mmolL¢ 1) 从外部到核心(图 3 b)。 结果表明 直径为 10 毫米。 然而,随着骨料尺寸的增加,更多的核心 细胞被封闭并与开放环境隔离。 微传感器测量表明大聚集体 (500 mm) 表现出比 100 毫米骨料。 H2 浓度达到 5.3 mmolL¢ 1 在大集合的核心,而 O2 随着探头深度的增加,浓度降至零 增加到近 50 毫米(图 3 b)。 因此,一个最优 用于光生物 H2 生产的 SFD 高度依赖 在总规模上。
在密闭玻璃管中的实验表明, 总体而言,相对于 10 毫米和 500 毫米的骨料, 100 mm 聚集体(具有相同的初始总叶绿素 content) 表现出卓越的制氢能力 (支持信息,图 S9 和图 S10)。 这 10 毫米的骨料几乎不能产生 H2 或性能 氢化酶活性。 顶空 H2 的量 由 500 毫米骨料产生的少于那些 由 100 毫米骨料在不同时间段产生的 (支持信息,图 S9)。 虽然体外 500 mm 聚集体中的氢化酶活性较高, 500 mm 聚集体的体内氢化酶活性低于 100 mm 聚集体(图 2 c 和支持信息,图 S11)。 这很可能 由于内部光合作用的不利限制 PSII 产生电子,这证明了 测量 PSII 活动(支持信息,图 S12)。 这表明氢化酶和 PSII 是保证光生物 H2 生产的关键。 PSII 效率的检查表明 Fv/Fm 500 毫米聚集体的值仅为 0.1-0.3。 没有 充足的光合电子,大细胞中的核心细胞 聚合体不能通过氢化酶有效地产生 H2 尽管氢化酶的活性很高。
众所周知,利用有氧光合作用 在有氧条件下能产生H2的微生物 将是生物 H2 向前迈出的重要一步 生产。 [18] 然而,筛选仍然是一个巨大的挑战 对于具有显着氧耐受性的天然物种。 最近,黄等人。 鉴定了一种新的微藻菌株 (Chlorella vulgaris YSL01) 并且该菌株可以连续 产生高达 1.9 mLH2 (Lculture)¢ 1 与 5% O2 在 10% 二氧化碳。 [9b] 然而,这种 H2 的产生只发生在 人工的而不是自然的有氧环境。 在我们的 研究表明,聚集的绿藻(C. pyrenoidosa)可以 连续产生约 0.5 mLH2 (30 mLculture)¢ 1, 相当于约 17 mLH2 (Lculture)¢ 1 . 这个 自然条件下的价值约为 9 倍以上 改良条件下的普通小球藻 YSL01。 [9b] 我们的 研究表明,绿藻聚集体是 在曝光期间能够光合自养 H2 到连续照明,这提供了第一种情况 在自然有氧条件下持续产生光生物 H2。 这一成就对于促进 绿色能源发展。
在我们的尝试中,原位硅化可以诱导细胞 C. Pyrenoidosa 的聚集,因为细胞可以 由二氧化硅材料凝聚,这取决于细胞表面的仿生化学修饰。 这种化学品—— 材料细胞工程可以产生H2的新功能 通过新型细胞材料复合物生产 可行、廉价且有效。 更一般地说, 类似的基于化学材料的细胞修饰可能 扩展到其他微生物以诱导设计的 功能转换。 它进一步遵循生物矿化启发的策略,通过化学和 物质途径。
这项研究得到了基础研究的支持 中央高校资助项目(浙大校长项目) 和国家自然科学基金 (21471129 和 31370270)
[1] a) O. Kruse, B. Hankamer, Curr. Opin. Biotechnol. 2010, 21, 238 – 243; b) K. Srirangan, M. E. Pyne, C. P. Chou, Bioresour. Technol. 2011, 102, 8589 – 8604.
[2] a) J. A. Turner, Science 1999, 285, 687 – 689; b) J. R. Bartels, M. B. Pate, N. K. Olson, Int. J. Hydrogen Energy 2010, 35, 8371 – 8384.
[3] S. J. Burgess, B. Tamburic, F. Zemichael, K. Hellgardt, P. J. Nixon, Adv. Appl. Microbiol. 2011, 75, 71 – 110.
[4] a) A. Hemschemeier, T. Happe, Biochim. Biophys. Acta Bioenerg. 2011, 1807, 919 – 926; b) S. Grewe, M. Ballottari, M. Alcocer, C. DÏAndrea, O. Blifernez-Klassen, B. Hankamer, J. H. Mussgnug, R. Bassi, O. Kruse, Plant Cell 2014, 26, 1598 – 1611.
[5] H. Gaffron, J. Rubin, J. Gen. Physiol. 1942, 26, 219 – 240.
[6] S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong, T. Happe, Proc. Natl. Acad. Sci. USA 2009, 106, 17331 – 17336.
[7] a) A. Melis, L. Zhang, M. Forestier, M. L. Ghirardi, M. Seibert, Plant Physiol. 2000, 122, 127 – 133; b) M. L. Ghirardi, L. Zhang, J. W. Lee, T. Flynn, M. Seibert, E. Greenbaum, A. Melis, Trends Biotechnol. 2000, 18, 506 – 511.
[8] E. Eroglu, A. Melis, Bioresour. Technol. 2011, 102, 8403 – 8413.
[9] a) A. Bandyopadhyay, J. Stçckel, H. Min, L. A. Sherman, H. B. Pakrasi, Nat. Commun. 2010, 1, 139; b) J.-H. Hwang, H. C. Kim, J.-A. Choi, R. A. I. A. -Shanab, B. A. Dempsey, J. M. Regan, J. R. Kim, H. Song, I.-H. Nam, S.-N. Kim, W. Lee, D. Park, Y. Kim, J. Choi, M.-K. Ji, W. Jung, B. H. Jeon, Nat. Commun. 2014, 5, 3234; c) M. L. Ghirardi, J. Cohen, P. King, K. Schulten, K. Kim, M. Seibert, Proc. SPIE-Int. Soc. Opt. Eng. 2006, 6340, U257 – U262.
[10] a) T. K. Antal, T. E. Krendeleva, T. V. Laurinavichene, V. V. Makarova, M. L. Ghirardi, A. B. Rubin, A. A. Tsygankov, M. Seibert, Biochim. Biophys. Acta Bioenerg. 2003, 1607, 153 – 160; b) A. A. Volgusheva, V. E. Zagidullin, T. K. Antal, B. N. Korvatovsky, T. E. Krendeleva, V. Z. Paschenko, A. B. Rubin, Biochim. Biophys. Acta Bioenerg. 2007, 1767, 559 – 564.
[11] a) C. E. Hamm, R. Merkel, O. Springer, P. Jurkojc, C. Maier, K. Prechtel, V. Smetacek, Nature 2003, 421, 841 – 843; b) F. Nudelman, N. A. J. M. Sommerdijk, Angew. Chem. Int. Ed. 2012, 51, 6582 – 6596; Angew. Chem. 2012, 124, 6686 – 6700; c) E. Buerlein, Angew. Chem. Int. Ed. 2003, 42, 614 – 641; Angew. Chem. 2003, 115, 636 – 664.
[12] a) B. Wang, P. Liu, W. Jiang, H. Pan, X. Xu, R. Tang, Angew. Chem. Int. Ed. 2008, 47, 3560 – 3564; Angew. Chem. 2008, 120, 3616 – 3620; b) S. H. Yang, K.-B. Lee, B. Kong, J.-H. Kim, H.-S. Kim, I. S. Choi, Angew. Chem. Int. Ed. 2009, 48, 9160 – 9163; Angew. Chem. 2009, 121, 9324 – 9327; c) E. H. Ko, Y. Yoon, J. H. Park, S. H. Yang, D. Hong, K.-B. Lee, H. K. Shon, T. G. Lee, I. S. Choi, Angew. Chem. Int. Ed. 2013, 52, 12279 – 12282; Angew. Chem. 2013, 125, 12505 – 12508; d) G. Wang, X. Li, L. Mo, Z. Song, W. Chen, Y. Deng, H. Zhao, E. Qin, C. Qin, R. Tang, Angew. Chem. Int. Ed. 2012, 51, 10576 – 10579; Angew. Chem. 2012, 124, 10728 – 10731; e) W. Xiong, Z. Yang, H. Zhai, G. Wang, X. Xu, W. Ma, R. Tang, Chem. Commun. 2013, 49, 7525 – 7527.
[13] T. Hîbert, L. Boon-Brett, G. Black, U. Banach, Sens. Actuators B 2011, 157, 329 – 352.
[14] L. J. Iwuchukwu, M. Vaughn, N. Myers, H. OÏNeill, P. Frymier, B. D. Bruce, Nat. Nanotechnol. 2009, 5, 73 – 79.
[15] M. L. Ghirardi, S. Kosourov, A. Tsygankov, M. Seiber-t, Proceedings of the 2000 DOE Hydrogen Program Review (San Ramon, CA) 2000, pp. 1 – 13. NREL/CP-570-28890.
[16] F. Rappaport, B. A. Diner, Coord. Chem. Rev. 2008, 252, 259 – 272.
[17] A. Volgusheva, S. Styring, F. Mamedov, Proc. Natl. Acad. Sci. USA 2013, 110, 7223 – 7228.
[18] G. Goldet, A. F. Wait, J. A. Cracknell, K. A. Vincent, M. Ludwig, O. Lenz, B. Friedrich, F. A. Armstrong, J. Am. Chem. Soc. 2008, 130, 11106 – 11113.
